Breakthrough Speed. Boundless Exploration.
From de novo design to retrosynthesis route planning, AIDDISON™ empowers medicinal chemists to make data-driven decisions at every stage of drug discovery with unprecedented efficiency.
30+
YEARS OF EXPERIMENTAL TRAINING DATA
65B+
MOLECULES IN VIRTUAL CHEMICAL SPACE
99%
FASTER THAN TRADITIONAL SCREENING
Seamless AI for Small Molecule Design
See how AIDDISON™ brings clarity and efficiency to your drug discovery workflows.

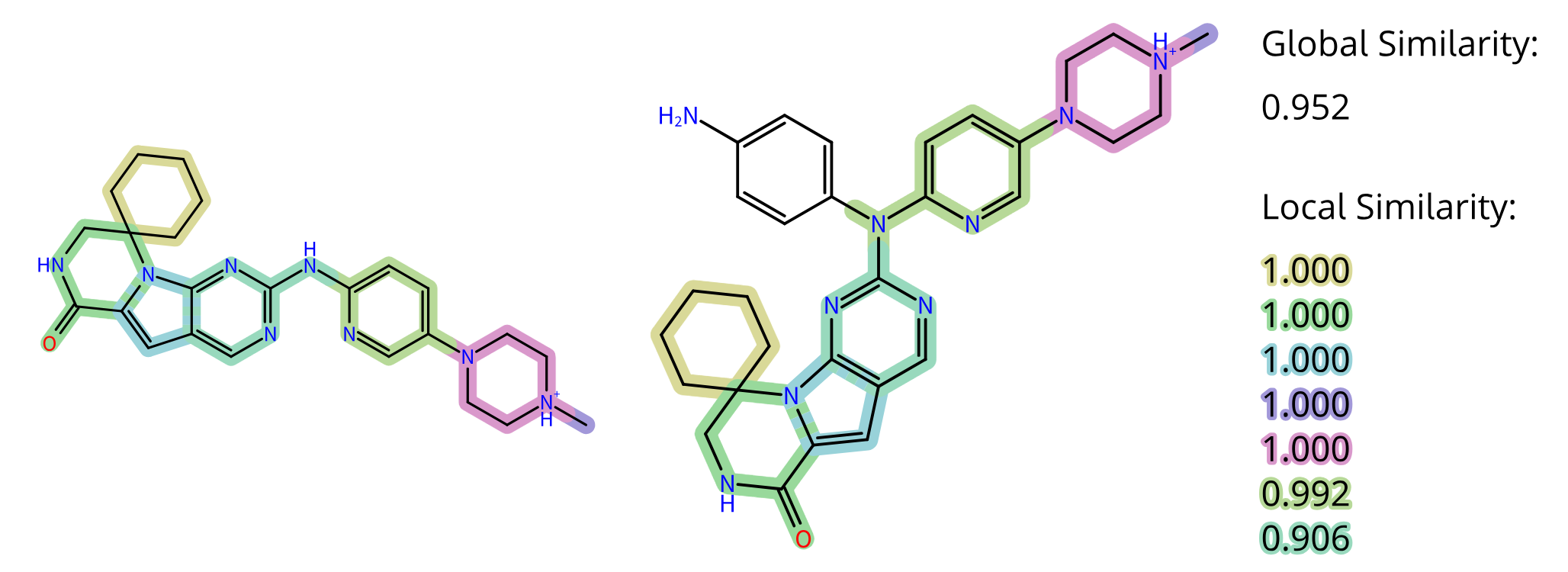
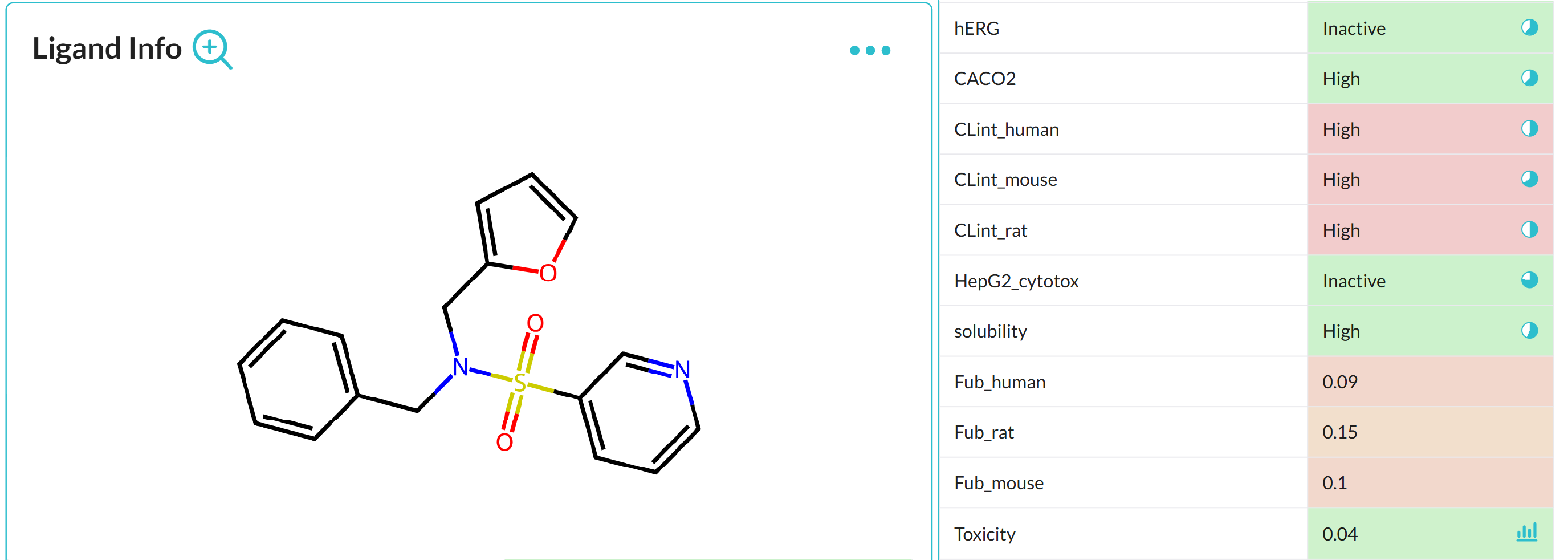
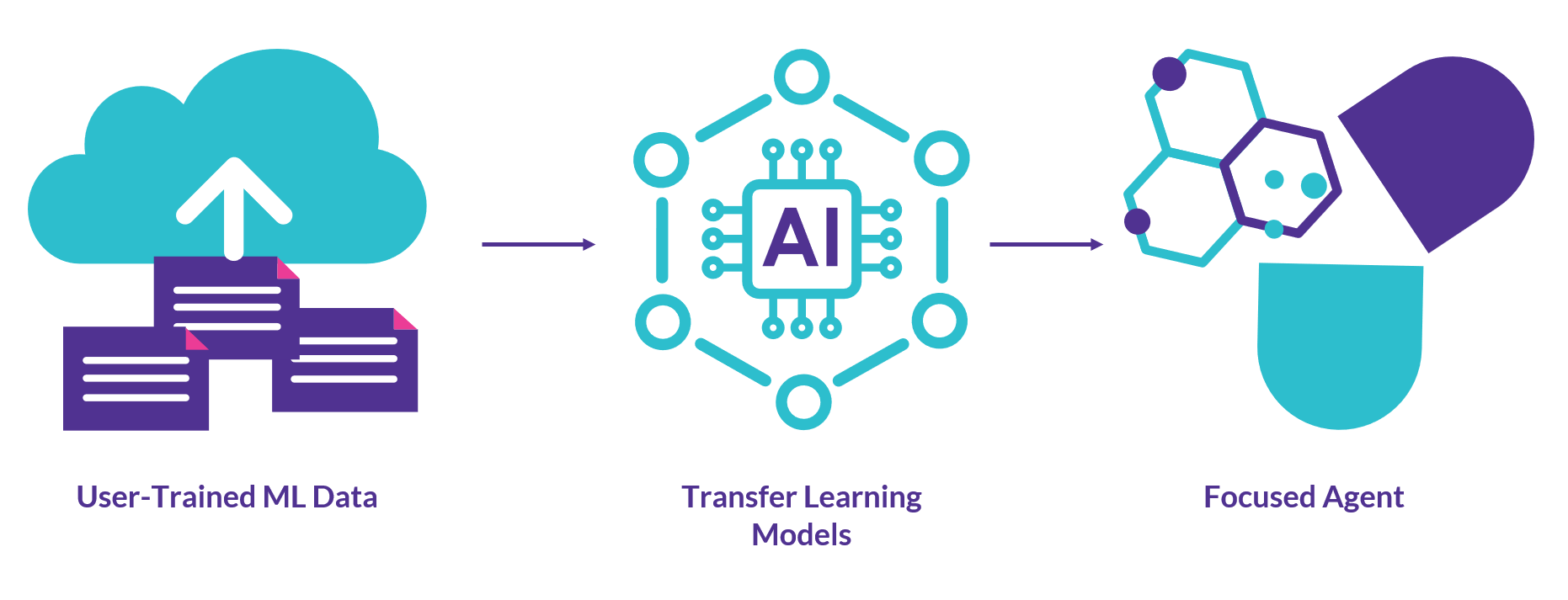
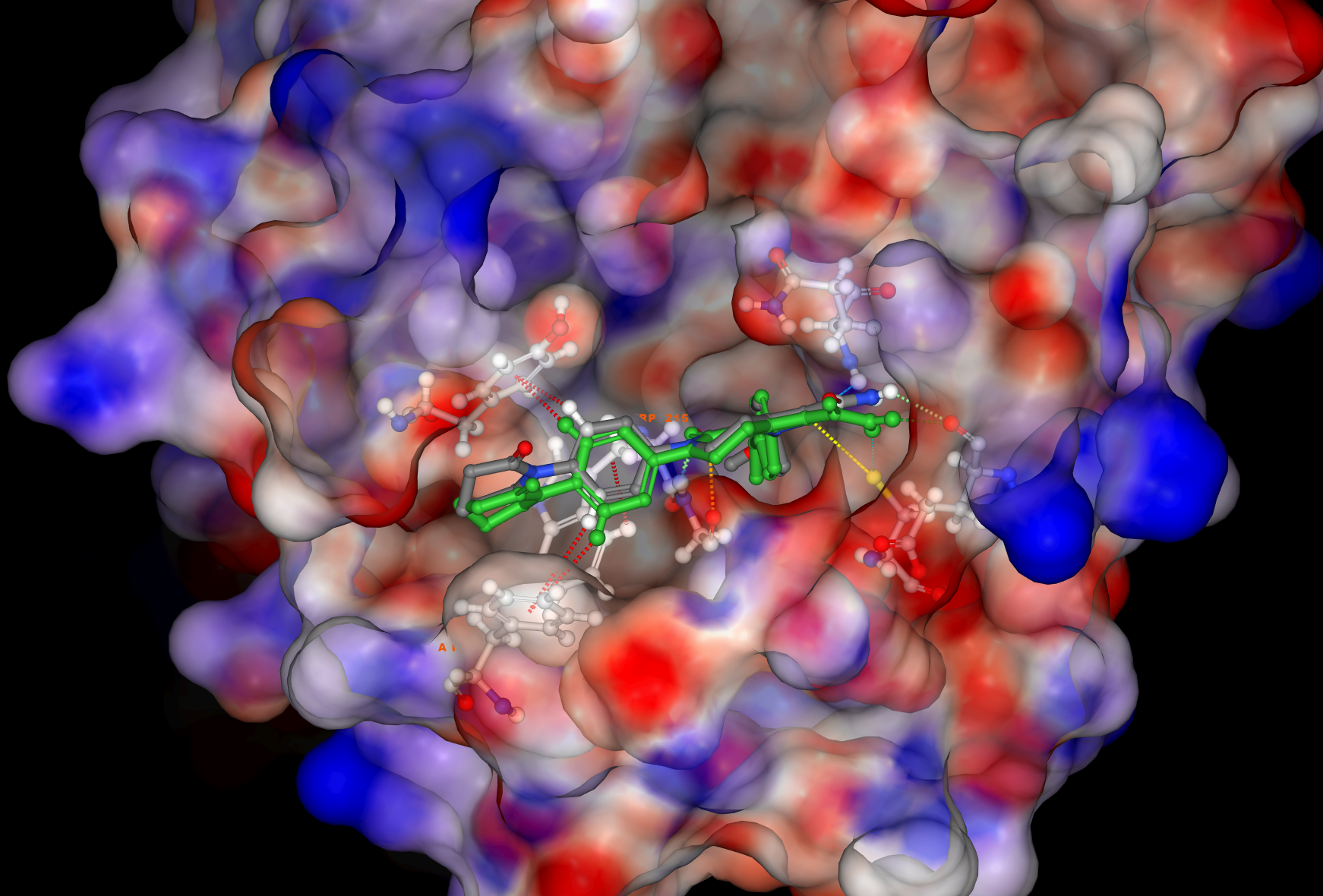
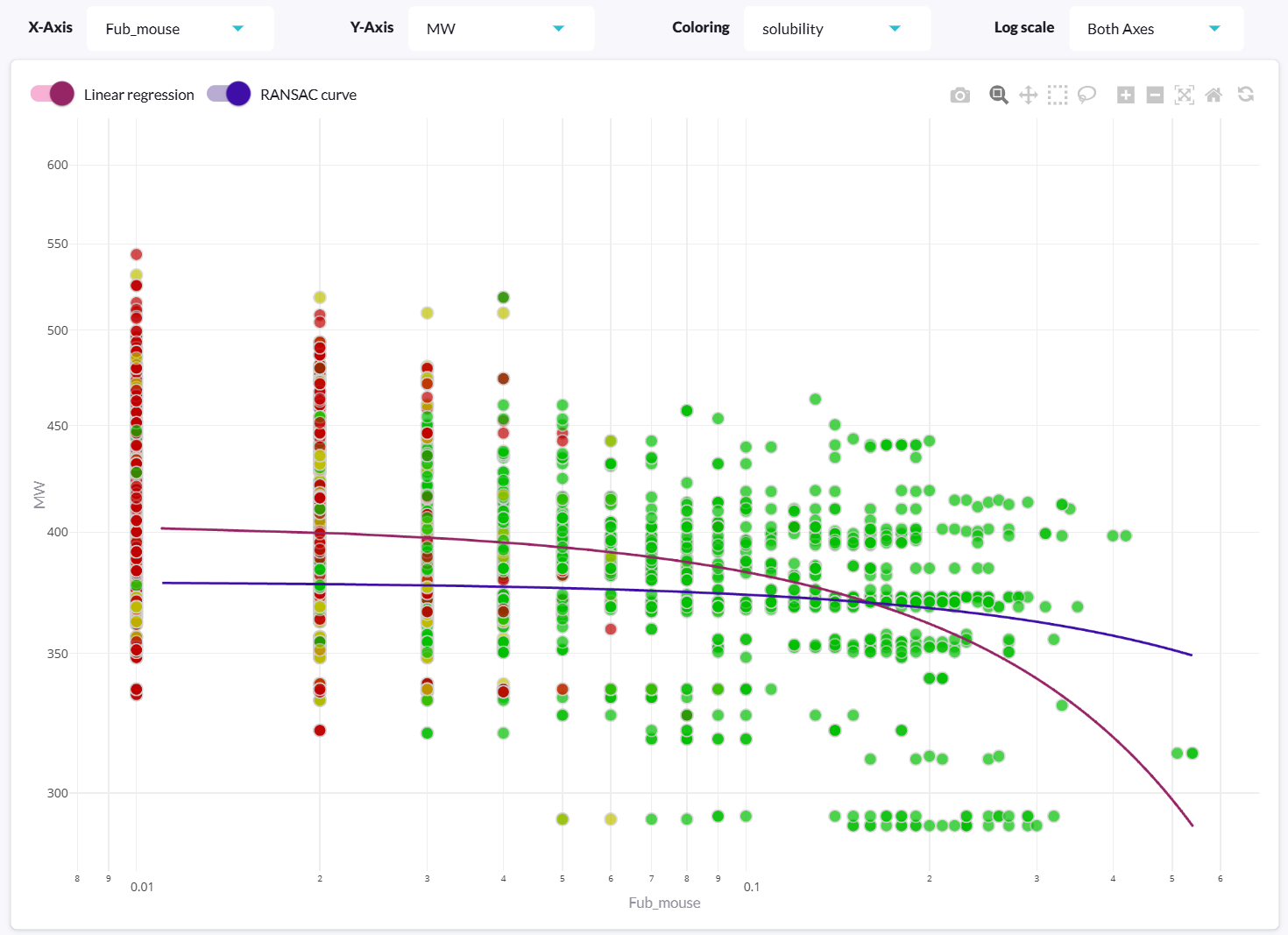
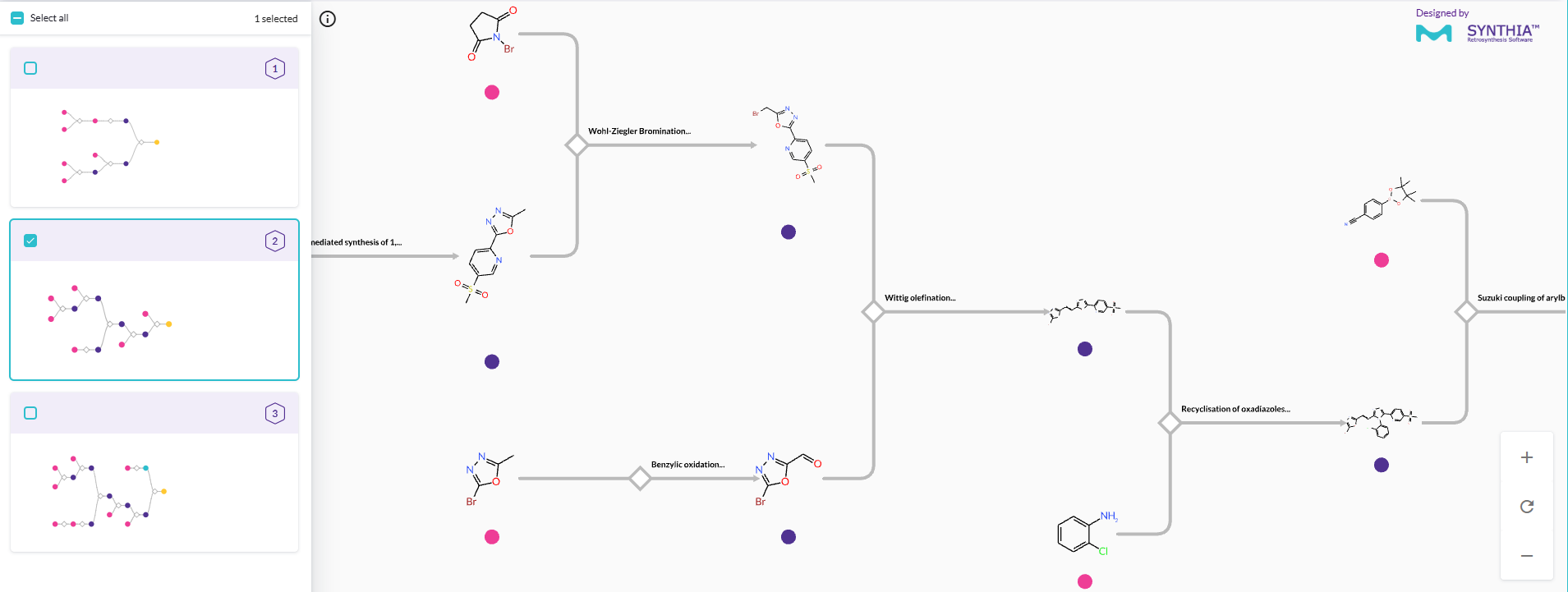

Benefits of aiddison™ IN DRUG DISCOVERY
All-in-One Drug Design
Virtually design, optimize, and plan synthesis—seamlessly in one platform.
Data-Driven Insights
Leverage 30+ years of real-world ADMET data to predict drug properties with confidence.
Designed for Efficiency
Navigate complex drug discovery with intuitive data visualization and explainable AI.
Streamlined Sourcing
Access our integrated chemistry platform to accelerate your journey from concept to clinic.
Licensing
One Platform. Endless Possibilities.
With AIDDISON™, you gain access to a suite of industry-leading tools, proprietary data models, and vast product catalogs—all integrated into a single, easy-to-use platform. Get in touch with our team to discuss flexible subscription options.


 1")
GET STARTED
Find the Right Molecule with AIDDISON™
Ready to see how AIDDISON™ can transform your drug discovery? Contact us today to learn more and request a demo of our solutions.
Resources
Explore our latest white papers, webinars, and more to stay informed about cutting-edge advancements in AI-powered drug discovery and retrosynthesis.

White Papers
Learn practical strategies for using AI/ML to find novel drug candidates and how to integrate AI tools into your existing workflows.

White Papers
This playbook provides a comprehensive guide to integrating AI into existing small molecule drug discovery programs.

Webinars
Discover how to apply generative AI methods in hit identification, hit-to-lead, and lead optimization of novel molecules.

